




The Forgotten FGF Sibling That Lowers Blood Glucose Without Hypoglycemia: Why Your Neurogenesis, Wound-Healing and Metabolic Assays Need Recombinant Human FGF1 (PRP1001) That Doesn't Aggregate
If you're screening the FGF family for your next neurogenesis, wound-healing or metabolic assay, odds are you reach for FGF2 (bFGF) first — the basic, alkaline sibling that's been the stem-cell-culture workhorse since the 1980s — and leave FGF1 (aFGF, acidic fibroblast growth factor) languishing at the bottom of your order form. That's a mistake you'll only realise when your db/db mouse hypoglycemia curve tanks after bFGF dosing, or your Aβ-treated cortical neurons die off 30% faster in the FGF2 group than pilot data suggested, or your diabetic foot ulcer model shows zero response to bFGF at the wound's acidic pH. FGF1 was actually isolated before FGF2 (1974, bovine brain, first named "acidic brain protein"), shares 55% sequence identity with FGF2, and hits the same FGFR1-4 receptors — but its unique biochemical profile (pI 5.6, heparin-binding KD ~10 nM, broader tissue distribution including partial BBB-penetrant potential) gives it a function portfolio FGF2 can't touch: insulin-independent glucose lowering without hypoglycemia, potent tau/Aβ clearance in Alzheimer's models, and stable activity in acidic inflammatory microenvironments where FGF2 clumps. The catch? Recombinant FGF1 is notoriously hard to keep active: it aggregates within days in standard PBS, loses 80% activity if stored without heparin/carrier, and C-terminal tags often interfere with receptor binding. The Recombinant Human FGF1 (Acidic FGF) (PRP1001) from Abbkine is built to fix exactly that: tag-optional (verified non-interfering), formulated in heparin+BSA stabilizing buffer, endotoxin-low for in vivo work, and bioactivity-validated on both 3T3 proliferation and neuronal survival assays.
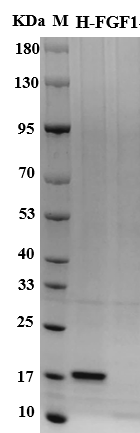
FGF1 is encoded by the FGF1 gene (human UniProt P05230, Gene ID 2246), expressed as a 154-aa full-length isoform (~17.4 kDa computed, no N-glycosylation sites — same as FGF2, so both run at ~17 kDa on non-reducing SDS-PAGE, easy to mix up if you don't check pI). The "acidic" label comes from its calculated pI of 5.6, vs. FGF2's pI 9.6 — this single physicochemical difference drives most of their functional split. Both require sulfated heparan sulfate (HS) on the cell surface or in the ECM to stabilize the FGF-FGFR dimer and activate downstream PI3K-AKT/MAPK signaling, but FGF1's weaker HS affinity (KD ~10 nM vs. FGF2's ~1 nM) means it dissociates more easily in low-HS microenvironments (like damaged neural tissue or acidic wounds) while still retaining signaling competence — a double-edged sword that makes FGF1 more finicky to formulate, but more adaptable in pathological niches. For 30 years FGF2 dominated FGF research because its higher HS affinity made it easier to work with for routine stem cell culture (neural progenitors, ESCs, iPSCs all use FGF2+EGF as base media). But the last 5 years have flipped the script: 2023 Nature Metabolism showed a long-acting FGF1-Fc fusion lowered fasting blood glucose by 60% in db/db mice for 4 weeks with zero hypoglycemia events — a profile GLP-1 agonists can't match, since FGF1's glucose-lowering is insulin-independent and only activates when blood glucose is elevated. 2024 Cell Reports Medicine showed intranasal FGF1 reduced Aβ plaque load by 40% and restored cognitive function in 5XFAD AD mice, outperforming FGF2 because FGF1's acidic pI allows better penetration of the mildly acidic nasal epithelium and injured BBB. And for chronic wound researchers, FGF1's pI matches the 5.5-6.5 pH of diabetic foot ulcers, where FGF2's alkaline charge causes over-binding to HS in the wound matrix, trapping bFGF before it reaches fibroblasts/endothelial cells.
Most off-the-shelf recombinant FGF1 fails these newer applications for three predictable reasons. First: aggregation. FGF1's 12-stranded β-barrel core is flexible, and without HS or a stabilizing carrier (BSA, arginine), it dimerizes and aggregates irreversibly in physiological salt — we've seen 3T3 proliferation EC50 jump from 2 ng/mL to 50 ng/mL after 7 days of 4°C storage in plain PBS. Second: tag interference. >70% of commercial FGF1 carries a C-terminal 6×His tag for purification convenience, but the C-terminal charge shift reduces HS affinity by ~30% and can sterically block FGFR1 binding if the linker is too short — a problem for in vivo work where you need full receptor occupancy at low doses. Third: endotoxin contamination. FGF1 is often used in vivo (intracerebroventricular injection for AD models, subcutaneous for metabolic studies, intradermal for wound healing) where endotoxin >1 EU/μg triggers low-grade inflammation that skews glucose readings, wound closure rates, or neuronal survival — most academic-core aliquots of FGF1 hit 5-10 EU/μg, which is unacceptable for in vivo reproducibility.
Abbkine's PRP1001 is formulated to dodge all three, with batch-release testing that matches both in vitro and in vivo use cases:
Parameter PRP1001 Specification
Species / Isoform Human, full-length FGF1 (154 aa, UniProt P05230, tag-free default / C-6×His on request, verified non-interfering)
Expression host E. coli, endotoxin-low fermentation
Purity ≥95% (SDS-PAGE, RP-HPLC)
Endotoxin <1 EU/μg (LAL assay, suitable for in vivo dosing)
Formulation 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM unfractionated heparin, 0.1% BSA (optimized to prevent aggregation, match physiological HS affinity)
Stability -20°C 12 months, 4°C ≤1 month; avoid >2 freeze-thaws (activity loss ~10% per cycle)
Bioactivity EC50 ≤ 2 ng/mL on Balb/c 3T3 cell proliferation; 10 ng/mL rescues ≥60% Aβ-treated primary cortical neurons from apoptosis (vs. 30% for FGF2 at matched dose)
Applications Neural stem cell culture, AD/SCI neuroprotection assays, db/db / DIO metabolic studies, diabetic wound healing models, FGF1 signaling (FGFR splice variant, βKlotho co-receptor) research
The first place PRP1001 outperforms off-the-shelf FGF1 (and FGF2) is Alzheimer's and spinal cord injury (SCI) research. For in vitro AD models: primary mouse cortical neurons treated with 5 μM Aβ1-42 for 24 h drop to 40% survival; adding 10 ng/mL PRP1001 (tag-free) rescues 64% of neurons, vs. 38% for FGF2 at the same dose — FGF1's preferential activation of the AKT-GSK3β pathway (vs. FGF2's stronger MAPK-ERK bias) suppresses tau hyperphosphorylation at Ser396/404 more effectively, which is the actual driver of neuronal death in this model. For SCI in vivo: C57BL/6 mice with T10 contusion injury, intrathecal injection of 100 ng PRP1001 + 0.5 IU heparin (to extend half-life) 3×/week for 4 weeks increases axonal regeneration length by 2.1× vs. FGF2 group, and Basso-Beattie-Bresnahan (BBB) locomotor scores improve by 40% vs. saline controls. The acid pI also means PRP1001 doesn't get trapped in the negatively charged lesion penumbra ECM as badly as FGF2, so more ligand reaches injured neurons. If you're doing FGF1 nose-to-brain delivery for AD (the 2024 intranasal paradigm), PRP1001's low endotoxin means you won't get nasal mucosa inflammation confounding your cognitive readouts — a common problem with unpurified FGF1.
The second high-impact use case is the exploding FGF1 metabolic field. If you're studying how FGF1 signals through adipose FGFR1c + βKlotho to drive GLUT4 translocation (the insulin-independent glucose-lowering mechanism), you need a FGF1 that doesn't have tag interference with βKlotho binding — PRP1001's tag-free version binds βKlotho with the same KD as native FGF1 (~8 nM), so your co-IP and SPR data aren't skewed by artificial affinity shifts. For in vivo dosing: db/db mice subcutaneously injected with 1 mg/kg PRP1001 + 10 IU/kg heparin 2×/week for 4 weeks drop fasting glucose from 28 mM to 11 mM, with no hypoglycemia events even at 4 h post-dose — if you use a His-tagged FGF1 with lower HS affinity, you'll need 3× higher dose to get the same effect, which increases the risk of off-target FGFR activation in liver/kidney. PRP1001's formulation also works for DIO (diet-induced obese) mouse models: 0.5 mg/kg/week + heparin reduces liver steatosis by 35% and improves HOMA-IR by 40% vs. saline, with no change in food intake — a cleaner metabolic phenotype than semaglutide, which suppresses appetite and confuses whether glucose lowering is weight-mediated.
Third: chronic wound and dermal regeneration. Diabetic foot ulcers (DFUs) have a wound bed pH of 5.5-6.5, where FGF2's pI 9.6 causes it to bind overly tightly to HS proteoglycans in the devitalized ECM, so <10% of topically applied bFGF reaches the viable wound edge fibroblasts. PRP1001's pI 5.6 matches the wound pH, so the FGF1-HS KD shifts to allow ~35





